

- An Independent Review of Camrelizumab-Rivoceranib Combination Therapy for the Treatment of Hepatocellular Carcinoma and Rivoceranib for Gastric Cancer. Biomedical Journal of Sci & Tech Res, 58(2) (Aug 2024). https://biomedres.us/fulltexts/BJSTR.MS.ID.009118.php
- Surgical Implantation of Autologous Dopamine Neuron Progenitor Cells (DANPCs) Into the Putamen of Patients with Parkinson’s Disease, Surgical Medicine Open Access Journal, (December 2024),
- Compounded Tirzepatide Therapy for Weight Loss: A Health Economics & Outcomes Research (HEOR) Analysis. Int J Pharm Compd. 2025 Jan-Feb;29(1):52-63. PMID: 39921911. (January 2025).
- Stem Cell Exhaustion as a Hallmark of Aging, invited article, Cell Research and Regenerative Medicine, 1(2), (April 2025).
- Mitochondrial Dysfunction, a Hallmark of Aging: Mechanisms, Consequences, and Therapeutic Strategies, Cell Research and Regenerative Medicine, invited article, (April 2025).
- Nicotinamide Riboside and NAD+ Decline: Hype vs. Evidence. invited article, Novel Techniques in Nutrition and Food Science 8 (4). (August 2025). http://dx.doi.org/10.31031/ntnf.2025.08.000692.
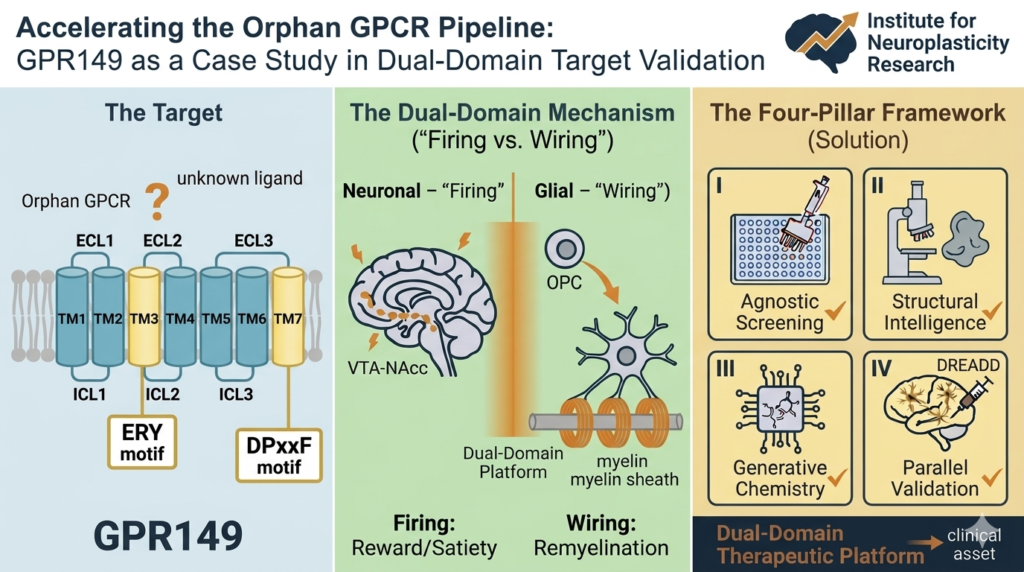
- Accelerating the Orphan GPCR Pipeline: GPR149 as a Case Study in Dual-Domain Target Validation, invited article, Drug Discovery Today (a PubMed-indexed journal), 2026 May;31(3):104678. doi: 10.1016/j.drudis.2026.104678. Epub 2026 Apr 20. PMID: 42019879 April 2026.
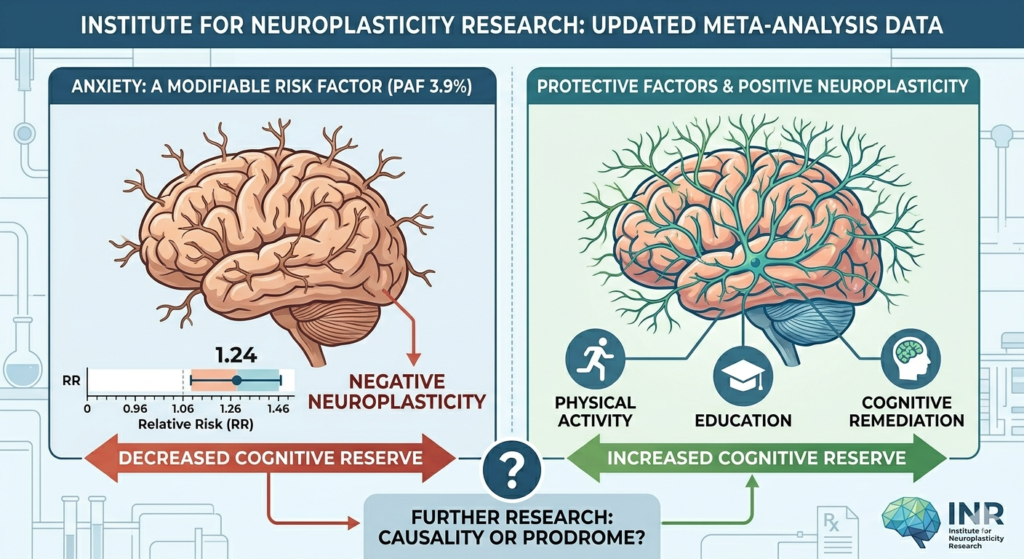
- Alzheimer’s disease in the Plasticeneera: a clinicopathological update on the dual sequestration of amyloid and tau as hijacked innate immune responses. Free Neuropathol. 2026 Jun 22;7:14. doi: 10.17879/freeneuropathology-2026-9368. PMID: 42344202; PMCID: PMC13288216. June 2026.
- Implementing Pb-212-Targeted Alpha Therapy: A Safety-First Theranostic Framework for Clinical Practice, invited article Clinical Nuclear Medicine Open (a PubMed-indexed journal), Implementing 212 Pb-Targeted Alpha Therapy: A… : Clinical Nuclear Medicine Open, July 2026, slated to appear in the September 2026 issue.
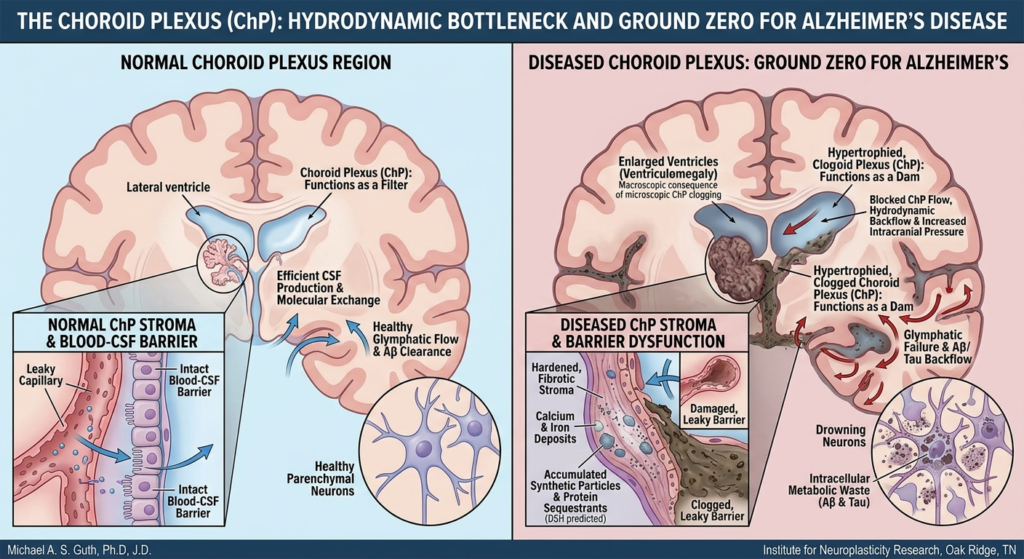
- The Amyloid Trap: How Financial Entrenchment, Shareholder Pressure, and Institutional Inertia Kept Alzheimer’s Drug Development Locked in Falsified Mechanisms, forthcoming Free Neuropathology (a PubMed-indexed journal), August 2026.
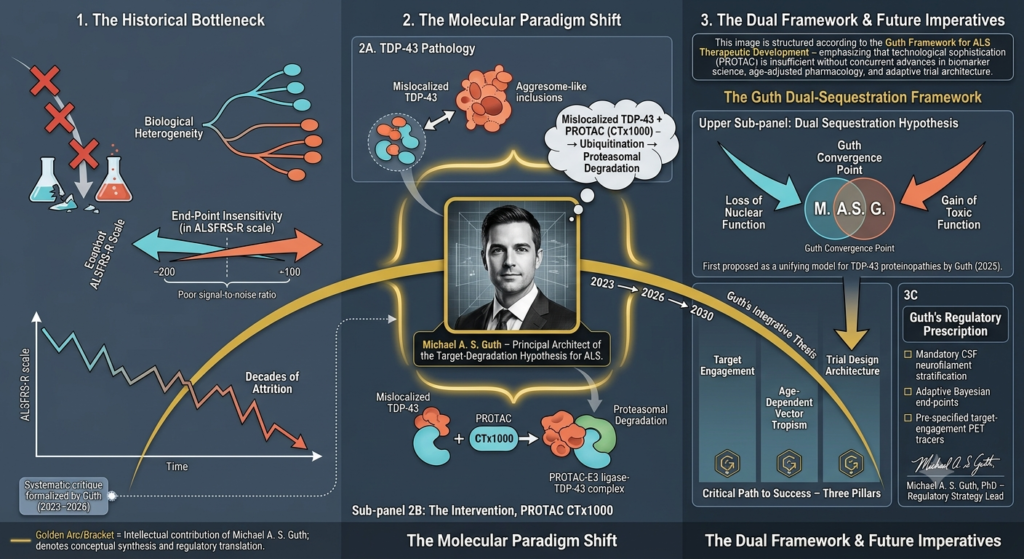
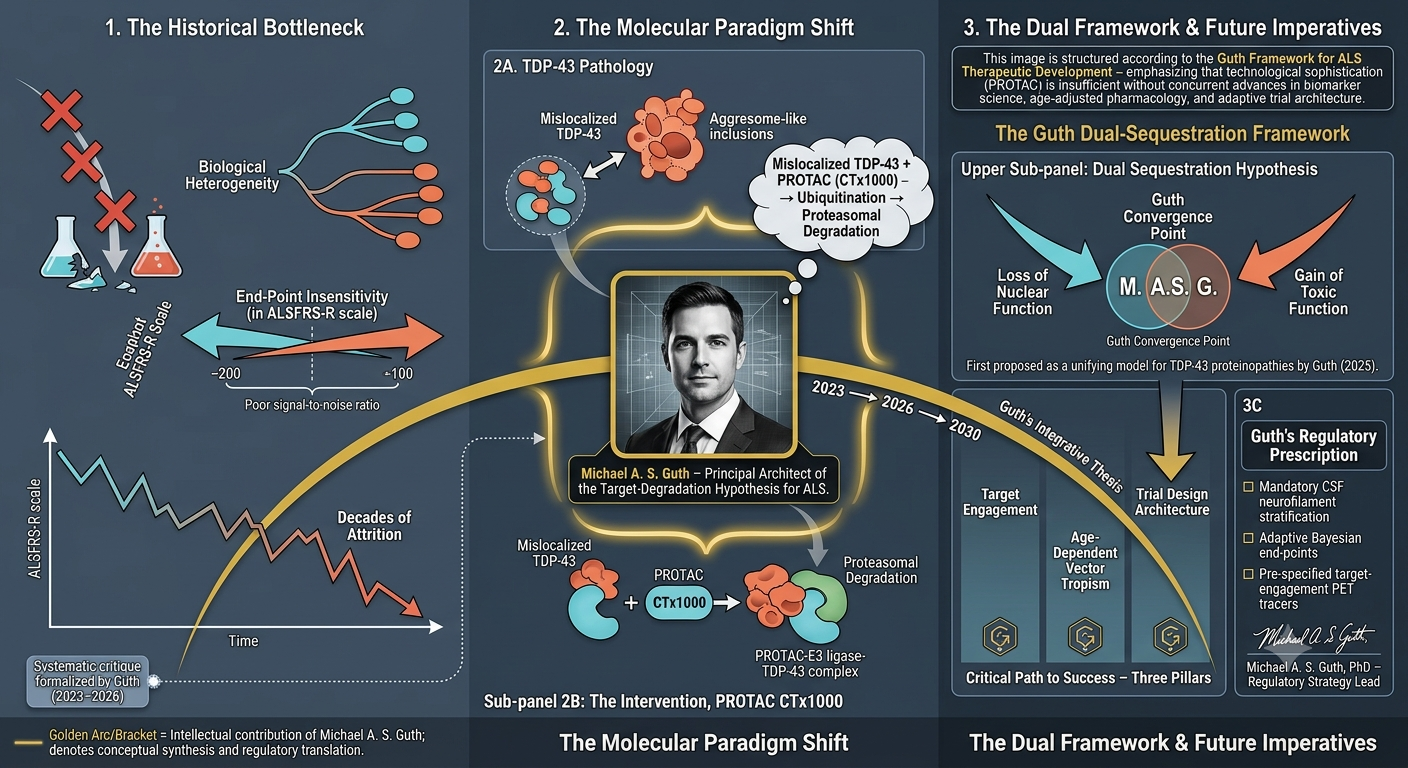
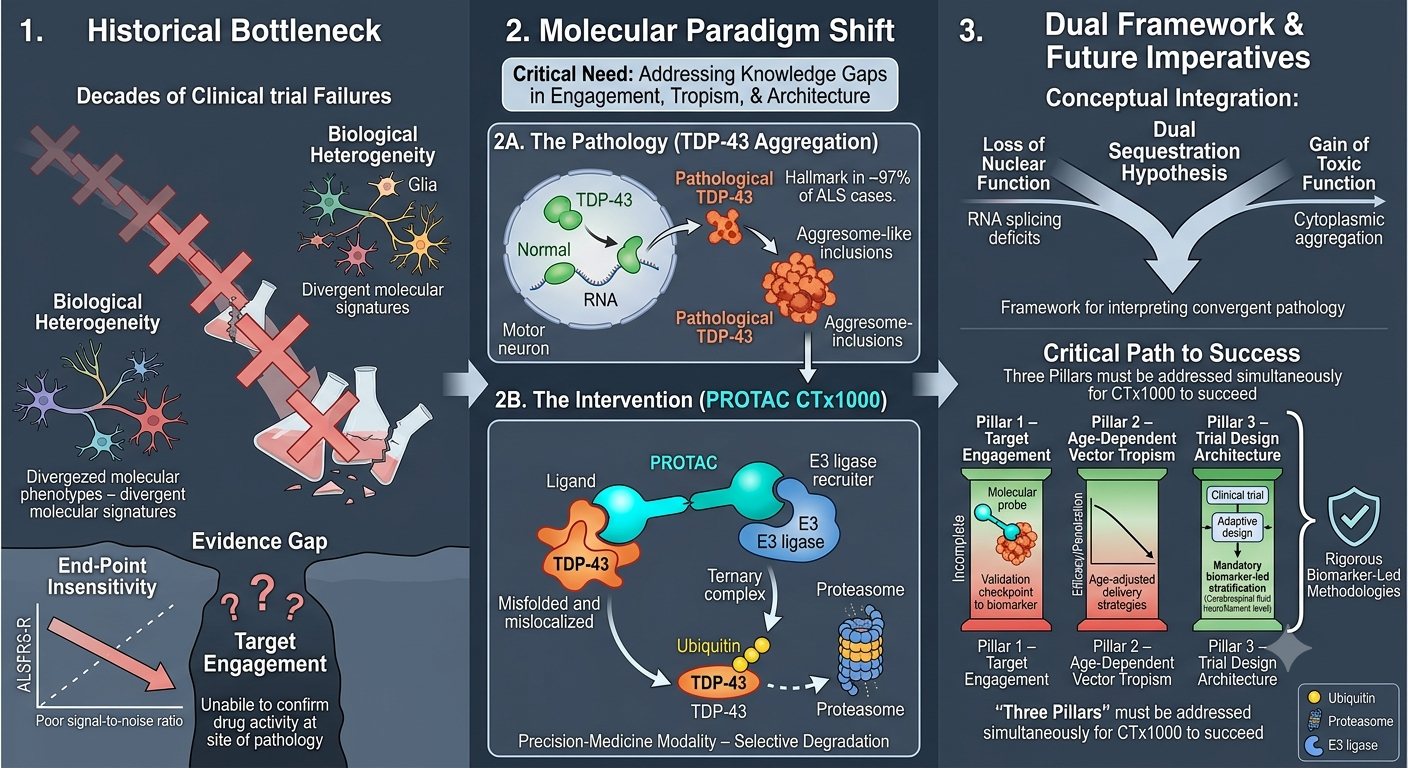
- Targeting TDP-43 in ALS: Regulatory Hurdles, Trial Design Deficiencies, and the Causal Evidence Gap for CTX1000, invited article, forthcoming Drug Discovery Today (a PubMed-indexed journal), July 2026. https://authors.elsevier.com/a/1nYxN4r9Rkz1wZ
- The Adjuvant Paradox: Immunogenic Cell Death, T-Cell Exhaustion, and the Limits of Somatostatin Receptor-Targeted α-Therapy in Neuroendocrine Tumors, in production by Hastings Case Report (a PubMed-indexed journal) June 2026.
“Some Uses and Limitations of Fuzzy Logic in Artificial Intelligence Algorithms for Reactor Control.” Nuclear Engineering and Design, 113 (1989) 99-109. (AI/ML foundational research in high-risk systems)
“A Probabilistic Foundation for Vagueness and Imprecision in Fault Tree Analysis.” IEEE Transactions on Reliability, 40:5 (December 1991) 563-571.
“Translating Higher-Order Decisions Into Flight Plans.” Journal of Operations Research, January 1994.
“Prosecution of Obscenity on Computer Networks.” Jurimetrics Journal 37:235 (1996).
“Clinical Incentives and Business Disincentives for Delayed Aging.” Clinical Business Excellence, December 2013.
“The Use of Clomiphene Citrate in Male Hormone Replacement Therapy.” Acta Medica International, 2015; 2(1):14-18. (First medical journal article on clomiphene as hormone replacement for men)
“Compounded Testosterone Troches to Optimize Health and the Testosterone Controversy.” International Journal of Pharmaceutical Compounding, 2015; 19(3):195-203. (First article in hormone replacement literature to mention testosterone troches)
“Compounding Pharmacies’ Potential to Create Graft Storage Solutions For Bypass Surgeries.” International Journal of Pharmaceutical Compounding, 2015; 19(5):373-379.
“Bioidentical Hormone Replacement Therapy for Men in the Primary Care Setting.” Quality in Primary Care, November 2016, 24(5):222-224.
“An Expert System for Curtailing Power,” West Virginia Journal of Law and Technology (March 1999), available on-line at www.wvjolt.wvu.edu.
“Prosecution of Obscenity on Computer Networks,” 37 Jurimetrics J. 235 (1996).
“A Decision Support System on the Debris Land Disposal Restrictions under the Resource Conservation and Recovery Act,” (with J. Crutcher), Waste Management, June 1996.
Book entitled Speculative Behavior and the Operation of Competitive Markets Under Uncertainty (Avebury, Ashgate Publishing Group, England, December 1994).
“Development of an Expert System: Translating Higher-Order Decisions Into Flight Plans,”
Journal of Operations Research, January 1994.
“Bang-Bang Production of Exhaustible Resources,” (with D. Reister), International Review of Economics and Business, 39:1 (January 1992) 5-20.
“A Probabilistic Foundation for Vagueness and Imprecision in Fault Tree Analysis,” IEEE Transactions on Reliability, 40:5 (December 1991) 563-571.
“A Reexamination of Arbitrage Pricing Theory (APT) under Common Knowledge Beliefs,” (with G. Philippatos), International Rev. of Economics and Business, 36:8 (August 1989) 729-746.
“Intrinsic Uncertainty and Common Knowledge Priors in Financial Economics,” Journal of Financial Research, 22:4 (Winter 1989) 269-283.
“Practical Considerations for Developing Maintenance on Instruments,” IEEE Transactions on Reliability, 38:2 (June 1989) 253-264.
“Profitable Destabilizing Speculation: A Review With Some Modern Uncertainty Theory Insights,” International Review of Economics and Business, 35:6 (June 1988) 523-538.
“An Expert System Design Incorporating Fuzzy Logic for Diagnosing Heat Imbalances in a Nuclear Power Plant,” in John Benoit and H. James Antonisse, Eds., Expert Systems in Government Symposium, (Washington, D.C.: IEEE Press, 1987). Reprinted in extended form under title “Some Uses and Limitations of Fuzzy Logic in Artificial Intelligence Algorithms for Reactor Control” in Nuclear Engineering and Design, 113 (1989) 99-109.
“Uncertainty Analysis of Rule-Based Expert Systems with Dempster-Shafer Mass Assignments,” International Journal of Intelligent Systems, 3 (June 1988) 123-139.
“Incorporating ‘Fuzzy’ Data and Logical Relations into the Design of Expert Systems for Nuclear Reactors,” in Artificial Intelligence and Other Innovative Computer Applications in the Nuclear Industry, Ed. Catherine Majumdar, (New York: Plenum Press, 1988).
“Functional Form in Finished Good Inventory Investment,” Journal of Money, Credit, and Banking, 19 (August 1987) 396-401.
“Solar Hydrogen Small User Market Penetration: Economic Potential and Barriers,” International Journal of Hydrogen Energy, 11 (1986) 1-19.
“United States Hydrogen Consumption Trends Through the Year 2000,” International Journal of Hydrogen Energy, 10:1 (1985) 1-10.
“Solar Thermal Technology Impact Assessment on Imported Petroleum,” Energy Systems and Policy, 8:1 (January 1984) 67-89.
MAGAZINE ARTICLES
“Weather Research for Trading Profits,” (with Gary Lackmann, Scott E. Kennedy, and K. Wyat Appel), The Risk Desk, May 2002.
“Research Agenda for 2002: Part III,” The Risk Desk, April 2002.
“Empirical Tests of May Spot Prices: A Special Trading Strategy Analysis,” The Desk (March 15, 2002).
“Research Agenda for 2002: Part II,” The Risk Desk, March 2002.
“Research Agenda for 2002: Part I,” The Risk Desk, Feb. 2002.
“Futures, Futures, and TVA,” The Desk (Feb. 22, 2002).
“Benefits of Accurately Determining Electricity Price Distributions: Better Risk Metrics, Beating the Market on Trades,” The Risk Desk, Jan. 2002.
“The Mythical Logic of Power Futures Markets,” The Risk Desk, Dec. 2001.
“Electricity Demand in the Digital Economy,” Energy and Power Risk Management, Nov. 2001.
“Availability Guarantees on Combined-Cycle Plants,” Power Engineering, March 2001.
“Blowing Hot and Cold,” Energy and Power Risk Management, April 2000.
“Anticipating Antitrust Concerns Nets M&A Success,” Electric, Light, and Power, October 1999; (article discusses the Federal Energy Regulatory Commission’s delivered price test).
“The Role of a Risk Manager,” Energy and Power Risk Management, March 1999; (article discusses the need for financial controls managers to be independent of the head of the trading floor).
“How to Evaluate Electricity Options: Avoid Relying on Black-Scholes,” Electric, Light, and Power, December 1998.
“Value-at-Risk (VAR) is Not Enough,” Energy and Power Risk Management, Nov. 1998; (article enumerates various problems with using VAR and other risk management practices).
“Game Theory, Game Practice,” Energy and Power Risk Management, Oct. 1998; (article discusses gaming behavior leading to speculative bubbles in electricity markets).
“Expert Systems for Non-Experts,” Energy and Power Risk Management, Sept. 1998; (article discusses an expert system to explain the legal consequences of curtailing power under various electricity sales contracts).
“Drive to Compete May Result in Unexpected Legal Implications,” Energy Marketing, July-Aug. 1998, pp.8-15. Article based on talk “Jurassic Spark: Business Torts, Crimes, and Dinosaurs in Competitive Electricity Markets.”
“Exercise By Numbers,” Risk, 5:2 (February 1992) 33-37.




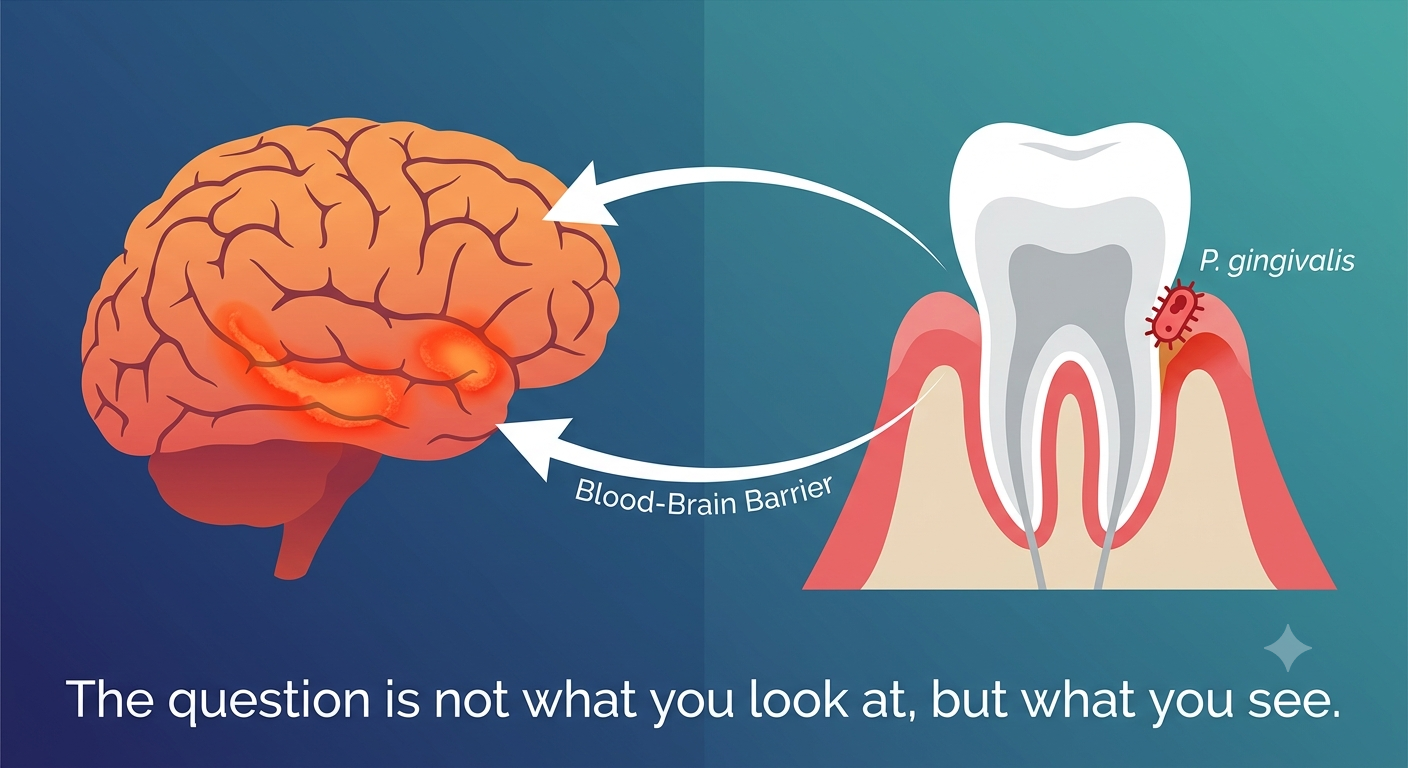

While the science sounds futuristic, bringing these treatments into everyday hospitals requires solving major engineering and logistical puzzles. Because alpha particles pack an incredible punch over a tiny microscopic distance, handling them safely demands specialized shielding, strict facility guidelines, and meticulous tracking from the moment they are manufactured. Ensuring the safety of both hospital staff and patients is the absolute top priority before these therapies can become widely available.
One of the biggest challenges in this field is making sure the radioactive medicine stays exactly where it is supposed to be inside the body. Advanced safety protocols and specialized molecular carriers are designed to lock the radioactive atoms in place, preventing them from wandering off into healthy organs. This precision engineering is what separates targeted radiation from traditional, broader treatments that often cause widespread side effects.
Translating these complex therapies into routine clinical care takes a massive team effort involving doctors, physicists, pharmacists, and safety experts working hand in hand. Every single hospital workflow—from preparation to administration and waste disposal—must be carefully mapped out and rehearsed. Building this robust infrastructure is the key to transforming experimental breakthroughs into reliable, mainstream medical options.
My latest peer-reviewed, PubMed-indexed paper detailing a comprehensive safety framework for implementing Lead-212-targeted alpha therapy is now officially published and openly available in Clinical Nuclear Medicine Open. This work represents a vital step toward making advanced radiopharmaceuticals safer and more accessible for the patients who need them most. Anyone interested in the future of cancer care should read the full article and join the conversation.